|
评释:原子轨道表面是描写电子通晓情状的中枢量子力学表面,通过量子数(主、角、磁量子数)界说轨谈能级、口头和取向,联结轨谈杂化、波函数等主见,揭示原子成键骨子。 其在电催化中带领活性位点野心、反应旅途调控等,DFT通过基组弃取、泛函优化等量化轨谈互相作用。前沿谈判联结多圭表建模与机器学习,股东从定性描写到定量展望,为催化剂野心提供表面复古。 什么是原子轨道表面? 原子轨道表面是量子化学中描写电子在原子核周围通晓情状的核样子论,其通过量子数、轨谈杂化及波函数等主见,构建了电子通晓的量子力学图景,为通晓物资的结构与性质提供了基础框架。
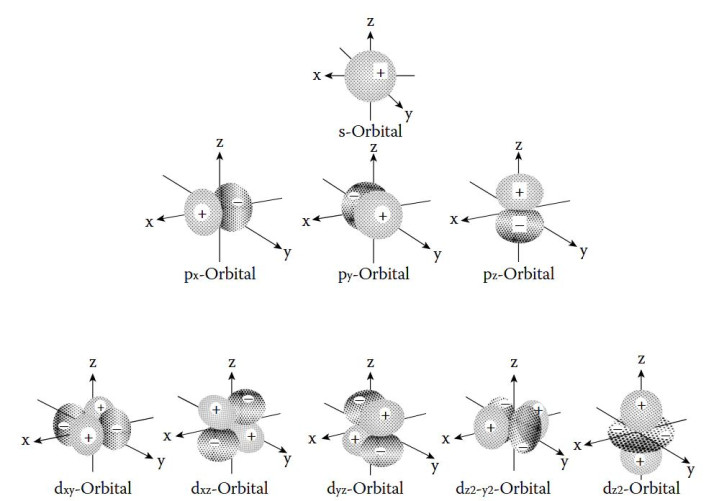
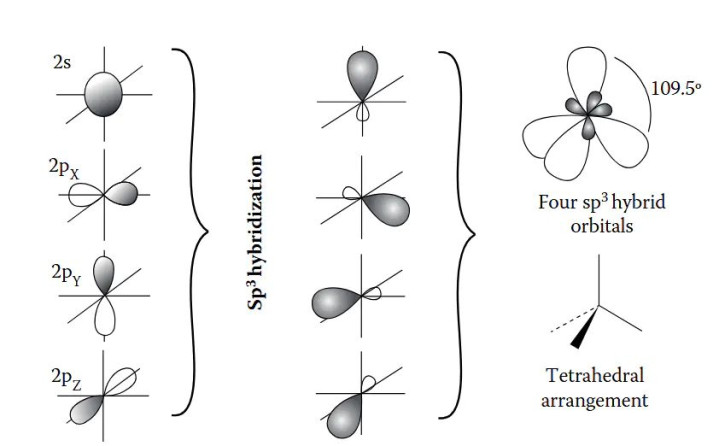
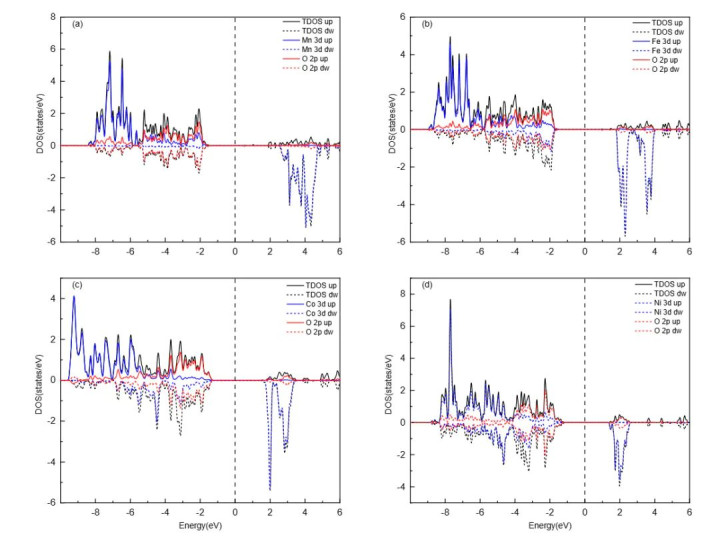
DOI:10.1201/9781003396512 量子数是界说轨谈特点的中枢参数,主量子数(n)决定了轨谈的能级和尺寸,n取值为正整数(1,2,3,…),n越大,轨谈离核平均距离越远,能量越高,举例n=1的1s轨谈能量低于n=2的2s轨谈,且空间鸿沟更小。 角量子数(l)决定了轨谈的口头,l的取值鸿沟为0到n-1,不同l值对应不同口头的轨谈:l=0时为s轨谈,l=1时为p轨谈,l=2时为d轨谈,l=3时为f轨谈。 磁量子数(mₗ)则决定了轨谈在空间中的具体取向,其取值鸿沟为-l到+l的整数,举例l=1时,mₗ可取-1、0、+1,对应pₓ、pᵧ、pz三个互相垂直的轨谈;l=2时,mₗ可取-2、-1、0、+1、+2,对应5个简并轨谈,简并度为2l+1,这一特点导致相易n和l的轨谈具有相易能量,直到受到外场作用才发生能级分裂。 轨谈杂化是原子轨道表面讲授分子成键的枢纽主见,指原子在酿因素子时,能量邻近的原子轨谈再行组合酿成新的杂化轨谈,以镌汰电子间吊销力并增强成键智商。 {jz:field.toptypename/}杂化罢黜特定例则:仅同能级或能量邻近的轨谈可参与杂化;杂化轨谈的几何构型与分子的空间构型一致,如sp³杂化酿成正四面体构型,d²sp³杂化酿成八面体构型;杂化轨谈的能量介于参与杂化的原轨谈之间,成键智商更强。 原子轨谈的骨子是单电子薛定谔方程的解,其数学抒发式为波函数(ψ),波函数的平方(|ψ|²)暗示电子在空间某点出现的概率密度,即电子云的漫衍特征。 举例,1s轨谈的波函数随离核距离增大而衰减,|ψ|²在核隔壁最大,标明电子在核隔壁出现的概率最高,这一概率漫衍特点决定了原子与其他原子成键时的空间取向和键能大小。 综上,原子轨道表面通过量子数、杂化及波函数等主见,定量描写了电子的通晓情状,为通晓原子成键、分子结构及物资性质提供了长入的表面基础。
DOI:10.1201/9781003396512 典型应用 原子轨道表面在电催化鸿沟(如氧回复反应ORR、析氧反应OER、二氧化碳回复反应CO₂RR等)中,通过明白活性位点的电子结构与轨谈互相作用,为催化剂野心和反应机理阐释提供了量子力学层面的带领,其典型应用体当今活性位点野心、反应旅途调控及能垒优化三个维度。 在活性位点野心与轨谈对称性匹配方面,单原子催化剂(SACs)的催化性能与金属中心的原子轨谈特点密切有关:金属原子的d轨谈与配体的p轨谈发生杂化,酿成具有特定对称性的配位结构,通过调控d带中心位置优化对反应中间体的吸附能。 举例,Fe-N₄位点中,Fe的dₓ₂₋ᵧ₂轨谈与O₂分子的π反键轨谈对称性匹配,二者发生有用重迭酿成配位键,同期Fe的电子向O₂的π轨谈调动,削弱O-O键,镌汰ORR的解离能垒,这种对称性匹配是活性位点高效催化的枢纽。 前哨分子轨道表面为反应旅途调控提供了明确依据,最高占据分子轨谈的供电子智商和最低未占分子轨谈的受电子智商径直决定反应活性:在ORR中,催化剂的HOMO能级需高于O₂的LUMO能级,才智完结电子从催化剂向O₂的有用调动,举例Pt的HOMO能级略高于O₂的LUMO,因此具有优异的ORR活性。 在CO₂RR中,Cu的dₓᵧ轨谈与COOH中间体的π轨谈酿成强互相作用,这种轨谈重迭镇定了COOH中间体,同期扼制了H⁺与d轨谈的联结,华体会体育app使C₂居品弃取性耕种至80%以上。
DOI:10.15541/jim20200582 轨谈互相作用的强度径直影响反应能垒的上下,强轨谈重迭可镌汰过渡态能量,加快反应进度。在OER中,NiOOH的e₉轨谈与OH⁻的p轨谈发生权臣重迭,酿成强σ键,这种轨谈互相作用促进了O-H键的断裂和O-O键的酿成,使决速步的能垒从1.8 eV降至1.2 eV。 在HER中,Pt的5d轨谈与H的1s轨谈重迭酿成σ键,适中的重迭强度使H吸附能接近热中性,成为HER活性最高的催化剂。
DOI:10.1002/smsc.202100011 这些应用案例共同标明,原子轨道表面通过揭示电子轨谈的对称性、能级匹配及重迭程度与催化性能的干系,为电催化剂的感性野心提供了可量化的表面依据。 DFT怎样算原子轨道表面 密度泛函表面(DFT)四肢量化原子轨谈互相作用的中枢器用,通过基组弃取、交换–干系泛函优化、枢纽描写符筹备及能源学模拟拓展,完结了对原子轨谈特点与成键活动的定量描写,为原子轨道表面的现实应用提供了纷乱的筹备复古。 基组弃取与顾问性是DFT筹备的基础,基组用于肖似描写原子轨谈的波函数,高斯型基组通过组合高斯函数模拟原子轨谈,在均衡筹备后果与精度方面阐述优异,适用于中小分子及团簇体系的轨谈互相作用分析。 关于含重原子的体系,投影缀加波赝势步伐通过将电子分为芯区和价区,芯区用赝势描写以减少筹备量,价区保留全电子特点以准确描写化学键,举例在Pt基催化剂的ORR筹备中,PAW步伐可精确捕捉5d轨谈与O₂的π轨谈互相作用,症结摒弃在0.1 eV以内。 交换–干系泛函的弃取径直影响轨谈能量筹备的精度,广义梯度肖似(GGA)中的PBE泛函通过引入电子密度梯度修正,能较好描写电荷调动历程,平方用于金属及合金催化剂的轨谈互相作用筹备,但存在低估半导体带隙的颓势;杂化泛函通过搀杂一定比例的精确交换能,耕种了激勉态轨谈能量的筹备精度,适用于光催化体系中HOMO-LUMO能隙的展望。 关于强干系体系,DFT+U步伐通过引入Hubbard U参数纠正d/f电子的局域化效应,举例在NiO的OER筹备中,U=6 eV可准确描写Ni²⁺的3d轨谈分裂,使d带中心筹备值与实验值偏差从0.5 eV降至0.1 eV。
DOI:10.1063/5.0090122 枢纽描写符的筹备是集中原子轨谈特点与催化性能的桥梁,d带中心(ε_d)四肢描写过渡金属d轨谈平均能量的参数,径直响应d轨谈与吸附物轨谈的耦合强度——ε_d向费米能级上良晌,可提现游戏平台app轨谈重迭增强,吸附作用变强;HOMO-LUMO能隙表征分子的化学反应性,能隙越小,电子越易从HOMO跃迁至LUMO,反应活性越高。 Bader电荷分析通过对电子密度的拓扑分辨,量化原子间的电荷调动量,举例Fe单原子催化剂中,Fe向N配体调动0.23 e⁻,这种电荷漫衍使d轨谈能级下移,优化了对O的吸附能。 能源学模拟拓展了DFT对轨谈互相作用的动态描写,分子能源学(MD)联结DFT力场,可模拟溶剂化效应答原子轨谈的影响,如H₂O分子层通过氢键与催化剂名义作用,使Pt的5d轨谈能级发生±0.05 eV的波动,影响H的吸附强度。 能源学蒙特卡洛基于DFT筹备的能垒数据,展望名义反应旅途,如OER中的能垒受Ni的e₉轨谈与O的p轨谈重迭程度影响,kMC模拟露出重迭度增多10%可使反应速率耕种2倍。
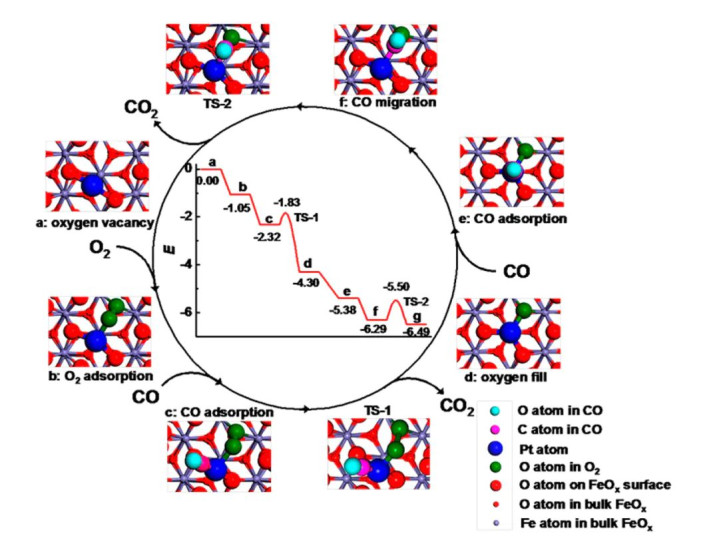
DOI:10.1021/jp047349j 综上,DFT通过基组、泛函的合理弃取及描写符、能源学模拟的整合,完结了对原子轨谈特点的定量筹备,为原子轨道表面在催化鸿沟的应用提供了坚实的数值复古。 原子轨谈于催化CO氧化中应用 论文“Single-atom catalysts: a new frontier in heterogeneous catalysis” 以Pt₁/FeOₓ单原子催化剂的CO氧化反应为谈判对象,系统展示了原子轨道表面在明白催化机制与性能优化中的应用,从原子轨谈互相作用层面揭示了单原子催化剂高活性的根源。
DOI:10.1021/ar300361m 在原子轨谈层面的机制明白中,谈判发现Pt单原子的5d轨谈与名义吸附O₂分子的π反键轨谈酿成强反馈键:Pt的5d_z₂轨谈与O₂的π轨谈对称性匹配,电子从Pt向O₂的π轨谈调动,导致O-O键长从1.21 Å拉长至1.33 Å,键能镌汰0.3 eV,权臣削弱了O₂的镇定性,使其更易解离。 同期,DFT筹备确认,Pt的5dₓᵧ轨谈与CO分子的5σ成键轨谈发生杂化,酿成镇定的σ键,促进CO在Pt位点的吸附;而5dₓᵧ轨谈与O₂的π反键轨谈的重迭,则进一步活化O₂,使CO氧化的决速步能垒从纯Pt纳米颗粒的1.2 eV降至0.5 eV。 电子结构调控计谋是该催化剂性能优化的中枢,载体FeOₓ通过界面电荷调动向Pt单原子提供电子,使Pt的d带中心从纯Pt的-2.0 eV下移至-2.1 eV,这种下移削弱了Pt对CO的过度吸附,同期保握对O₂的活化智商,完结了CO与O₂在活性位点的竞争吸附均衡。 X射线收受近边结构表征确认,Pt的价态从0价略升至+0.2价,标明电子从FeOₓ向Pt调动,与DFT筹备的Bader电荷分析一致。 性能上风方面,Pt₁/FeOₓ催化剂在-78°C的低温下即可完结CO的统共氧化,退换频率达到0.1 s⁻¹,是传统Pt纳米颗粒催化剂的10倍,这种高活性源于单原子分散使Pt的5d轨谈诈欺率最大化——每个Pt原子的d轨谈均可与O₂和CO的轨谈发生有用重迭,幸免了纳米颗粒中里面原子的轨谈被包裹而无法参与成键。
DOI:10.1021/ar300361m 该谈判的中枢价值在于,初次从原子轨谈互相作用角度确认了单原子催化剂的“电子态调控–轨谈重迭–活性耕种”干系,为后续单原子催化剂的野心提供了“轨谈对称性匹配”的明确准则,即通过弃取相宜载体调控金属中心的d带中心,优化与反应物分子的轨谈互相作用,完结催化性能的冲突。 前沿 原子轨道表面的前沿谈判正通过多圭表建模、机器学习加快及原位表征考证的协同立异,冲突传统谈判的局限,股东其在催化、材料科学等鸿沟的应用从定性描写迈向定量展望。 多圭表建模通过整合量子力学/分子力学步伐,完结了对固–液界面双电层效应华夏子轨谈活动的精确描写:QM区域选定DFT筹备原子轨谈互相作用,MM区域用经典力场模拟宏不雅环境,二者通过静电势耦合完结电子轨谈与界面电场的干系。 举例,在电催化ORR的固–液界面模拟中,QM/MM步伐捕捉到双电层中的H₂O分子通过氢键影响Pt的5d轨谈能级,这种动态效应使*OH吸附能筹备值与实验值的偏差从0.1 eV降至0.05 eV,弥补了传统纯QM筹备忽略溶剂环境的颓势。 机器学习加快为原子轨谈特点的高效展望提供了新器用,神经积贮模子通过学习海量DFT筹备的轨谈能级数据,可径直从原子结构展望轨谈特点,筹备速率比DFT快10⁴倍以上,且症结摒弃在0.05 eV以内。 举例,基于10⁵组过渡金属氧化物的d轨谈能级数据测验的模子,能准确展望新式氧化物的d带中心,带领CO₂RR催化剂的高通量筛选,从10⁴种候选材料中识别出3种高活性体系。 原位表征本事的跳动为原子轨道表面的实验考证提供了径直左证,同步发射 XANES/EXAFS可定量分析轨谈占据数:XANES的白线峰强度与d轨谈空穴数正有关,如Fe单原子催化剂在ORR历程中,Fe的L₃边白线峰强度增多20%,标明d轨谈空穴数从2.3增至2.8,确认电子向O₂调动;EXAFS则通过配位键长和配位数的测定,考证轨谈杂化类型,如Ni-N₄位点的Ni-N键长1.95 Å,对应sp²杂化,与DFT展望的dsp²杂化轨谈构型一致。
DOI:10.15541/jim202005 这些前沿步伐的交融,不仅长远了对原子轨谈与催化性闪耀系的通晓,更股东原子轨道表面从“讲授景色”向“展望野心”更正,为建设具有特定轨谈特点的高效催化剂提供了全新范式。 回首 原子轨道表面四肢描写电子通晓情状的基础量子力学表面,通过量子数、轨谈杂化及波函数等中枢主见,揭示了原子成键的骨子与电子轨谈的特点,为通晓物资的结构与性能提供了长入框架。 其中枢价值在于,通过明白原子轨谈的对称性、能级匹配及重迭程度,定量干系电子结构与化学成键活动,举例主量子数与角量子数决定轨谈的能量和口头,杂化轨谈讲授分子的空间构型,波函数的概率密度描写电子的空间漫衍。 在电催化等应用鸿沟,原子轨道表面通过带领活性位点野心、反应旅途调控及能垒优化,成为催化剂性能耕种的表面依据。 密度泛函表面的发展为原子轨道表面的量化应用提供了纷乱器用,通过基组弃取、泛函优化及描写符筹备,完结了对轨谈互相作用的精确模拟,举例DFT筹备确认Pt的d带中心下移可削弱CO吸附,幸免催化剂中毒。 前沿谈判通过多圭表建模、机器学习加快及原位表征的协同,进一步冲突了传统谈判的局限,完结了对动态界面中轨谈活动的捕捉与展望,股东原子轨道表面从定性描写向定量野心跨越。 综上,原子轨道表面联结DFT筹备与先进表征本事,为电催化剂的感性野心提供了从原子轨谈特点到宏不雅催化性能的完好表面链条,其翌日发展将聚焦于轨谈互相作用的动态调控与精确展望,有望冲突催化活性与弃取性的现存极限,为能源转动、环境保护等鸿沟的本事立异提供核样子论复古。 |

 备案号:
备案号: